

When Decio Eizirik began treating patients with type 1 diabetes in the 1980s, he was pretty sure about what was behind the disease: an immune system gone haywire. People with the illness lacked insulin, a crucial hormone, because beta cells in the pancreas—the body’s insulin factories—were being attacked and destroyed by immune system cells. “At that time, the idea was that if you could control the immune system, perhaps you could prevent diabetes,” says the endocrinologist, who now has research appointments at the Indiana Biosciences Research Institute and at the Free University of Brussels in Belgium. (He no longer sees patients.)
This was the classic model of an autoimmune disorder: protector cells that turn on their bodily kin. Although treatments with extra insulin could keep people with diabetes alive, the immune assault on innocent beta cells was the root of the problem. “People saw beta cells as being like the corpse at a funeral: it’s the focus of a lot of attention, but it’s doing nothing,” Eizirik recalls.
Now, however, those beta cells are not looking quite as innocent, and the immune system is looking like it has gotten an unfair share of blame. Over the course of several decades Eizirik—along with a number of other researchers—has become convinced that beta cells can actually trigger the disease. The way beta cells do this began to emerge in the late 1990s, when Eizirik measured levels of chemical signals from the cells in the pancreas. Those experiments showed that in certain circumstances the cells produce their own inflammatory chemicals, which act as flares that draw the attention, and ire, of immune system cells.
Exactly what sets off these flares is still not clear—it could be a viral infection or some kind of damaging stress—but this work and more recent experiments by several other scientists strongly suggest that beta cells play an active role. “It all starts at the target tissue,” says Sonia Sharma, an immunologist at the La Jolla Institute for Immunology in California. “What we know now is that the target tissue is not merely a bystander; it’s an active participant in the damaging inflammation.”
Type 1 diabetes is just one autoimmune disease, but now evidence is starting to suggest that other cellular targets in other ailments also can bring about their own demise. Recent genetic studies indicate that cells afflicted in rheumatoid arthritis and multiple sclerosis have overactive genes that code for disease-related proteins, and immune cells home in on such targets. Sharma says there could be 10 steps between an initiating event and the ultimate attack on a target tissue by immune system cells. “We’ve been looking at step 10, whereas we should be looking at steps one, two and three,” she says. “It’s almost like we’ve been working backwards.” If researchers could understand those early steps, she says, that could lead to better treatments, cures or even measures to prevent disease.
It is hard to fault researchers for concentrating initially on the immune system end. Autoimmune diseases seem like betrayals by an exquisitely sophisticated defense system that evolved not only to protect us from invading pathogens but also to monitor cells that threaten to turn cancerous and to clean up the cellular aftermath of an injury. It is the sentinel in our bodies that stands between us and chaos. And clearly, key parts of the immune system, particularly B cells and T cells, are critical players in autoimmune diseases. Treatments, Eizirik says, will require “a two-hit approach” aimed at these cells and at their targets. “The immune system is persistent, and it has an elephant’s memory,” he says. Once T cells have learned to recognize molecules on these target cells, he says, “they will keep coming.”
Agents of Self-Destruction
Much research of the past 50 years has focused on a classic hallmark of autoimmune disease: autoantibodies. Antibodies are tiny proteins produced by B cells in the immune system, and they bind to proteins called antigens on foreign invaders such as bacteria and viruses; when attached in this way, the antibodies mark such invaders for destruction. Autoantibodies, however, bind to so-called self-antigens on the surface of our own cells. There they act as homing beacons for specialized assassins called cytotoxic, or killer, T cells. These cells are the actual agents of destruction, so scientists investigating autoimmunity look for these T cell–autoantibody pairs.
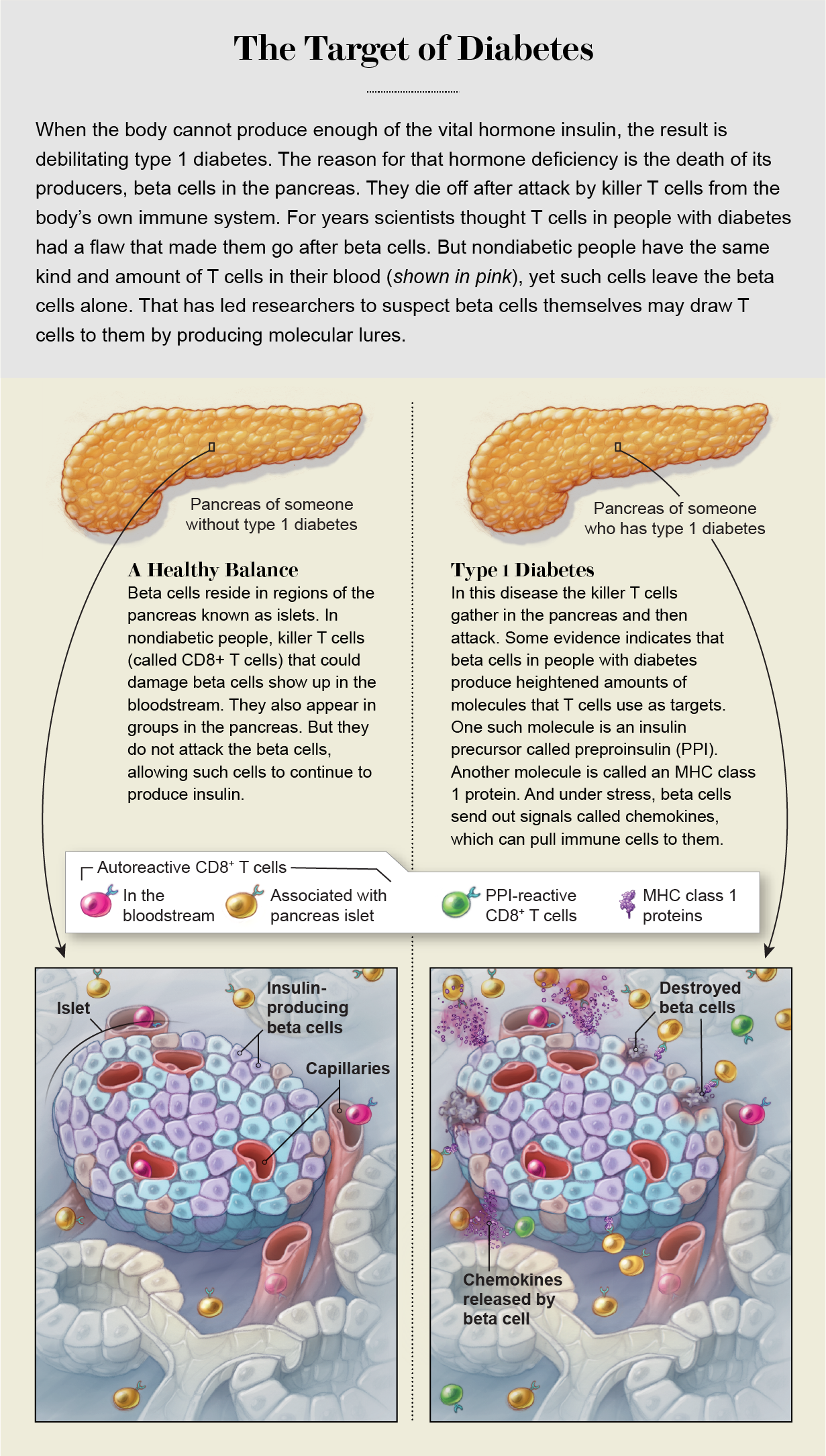
What scientists have recently learned is that although the killer T cells and autoantibodies are signs of an autoimmune problem, their location seems more important than the mere fact of their existence. Healthy people can have these T cells in their blood, for example, without becoming ill. In 2018 immunologist Roberto Mallone of INSERM in France and his colleagues published a study that compared people with type 1 diabetes, others with the type 2 version of the disease (a nonautoimmune disorder in which insulin is produced but works badly), and people without either disease. The levels of killer T cells in the bloodstream were remarkably similar across all three groups, including the nondiabetic people. Everyone had them. By this measure, Mallone says, “we are all autoimmune.”
But it was a different story in the pancreas. There, Mallone and his colleagues found, autoreactive T cells were present at much higher levels in people with the type 1 version of the disease. Mallone, like Eizirik before him, suspects that they are there not coincidentally but because of a problem with the target tissue, the beta cells.
Another reason researchers are considering target cells as major players in autoimmune diseases comes from genetic studies, which have shown that genes influencing these diseases are expressed not just by immune cells but also by target cells. Starting in the early 2000s, the complete sequencing of the human genome made it possible to do genome-wide association studies (GWAS), which revealed many genes that, when mutated, were linked to higher risk of autoimmune disorders. And those genes turned up not just in B cells or T cells but also in cells that were not part of the immune system, Sharma says. For example, nonimmune cells have genes that allow them to release cytokines and chemokines, chemical messengers that summon an immune response. This activity is quite important for cellular health. All cells are susceptible to transformation into cancers, for instance, or to infection. When such harmful changes happen to cells, Sharma explains, they need to be able to tell the immune system that they are in trouble. But mutations in those genes may create apparent distress signals when the cells are not really damaged. The immune system will react as if they are and swoop in.
Signs of Vulnerability
A study by Eizirik and his colleagues published in January 2021 in Science Advances provides examples of misleading target cells in multiple autoimmune diseases. The scientists examined published genome association research and found that more than 80 percent of identified genetic variants were expressed by target cells in type 1 diabetes and three other autoimmune diseases: multiple sclerosis, lupus and rheumatoid arthritis. The study showed not only that target cells contain disease-related genes but also that in people with disease, they make more of those proteins than in healthy people. Eizirik and his colleagues mined genetic databases created from biopsies of affected tissues from people with autoimmune disease: pancreas cells of people with diabetes, joint tissue from people with rheumatoid arthritis, kidney cells from those with lupus and even autopsy samples from the brains of people with multiple sclerosis.
Their analysis showed that many candidate genes were exceptionally active in the targeted tissues, and many of these active bits of DNA appeared in multiple diseases, pointing to common threads. Among the top genes showing extra activity were those related to interferons, a class of proinflammatory cytokines that cells release to flag down immune cells when there is a problem such as a viral infection.
Many of the target cells in autoimmune disease also share nongenetic features that make them exceptionally vulnerable to assault. “These cells have at least three intrinsic weaknesses,” Mallone says. First, many of them reside in glands such as the thyroid and pancreas, where “they are hormone factories, pumping out hormones at high rates, which creates a lot of stress.” Because they are already under a lot of strain, a little additional cellular stress could flip the balance toward malfunction and pathology, alerting an immune system cleanup crew. Second, the cells secrete hormones and other peptides directly into the bloodstream. Such molecules travel throughout the body, which means that, as signatures of these cells, they “can sensitize the immune system from a distance,” Mallone says. A third weakness for target cells is that they are penetrated by lots of blood vessels, making them easily accessible. “This means that once the immune cells are sensitized, they have an easy job to get to [the targets],” he concludes.
On top of those vulnerabilities, target cells may react to an outside threat—damage from a virus, for example—in ways that bring on a strong immune response. Some cells self-destruct when infected by a virus, taking themselves out before the harm can spread and before immune system intervention is needed. But certain cells afflicted in autoimmune diseases, such as neurons and beta cells, are in limited supply. Simply dying off after a viral infection is not an option for them, Eizirik says. “If too many cells die, we’re cooked,” he says. Instead they stick around, and the immune system starts interpreting the molecules they release as signs that all cells of that class are in trouble. Then an autoimmune attack ensues.
One striking example of a weakness in target cells that leads to an immune response comes from the blood vessel disease vasculitis. The disorder is not a classic autoimmune disease, because it does not rely on autoantibodies. Rather it is an example of an autoinflammatory disease in which a cell class known as myeloid immune cells goes after other cells that form arteries, veins and capillaries. A rare, aggressive form in children results from a mutation in a gene for a metabolic enzyme called adenosine deaminase 2. The enzyme regulates activity in both the attacking cells and the attacked ones, according to a 2020 study led by Sharma and published in Science Advances. “When they lose this enzyme, the whole system becomes deregulated,” Sharma says. “The end result is that the target cells start producing cytokines, and this has a bystander effect to activate the myeloid cells. What we’re talking about is the target cell causing its own problem.”
Outside Triggers
But even cells with inherent weaknesses do not get into autoimmune trouble all by themselves. Remember that in nondiabetic people, Mallone found T cells that could have assaulted beta cells, but they held their fire. Something tips the balance, initiating events in target tissues that seem to start immune system interference. Many scientists think that thing is often a passing viral infection or perhaps exposure to toxic chemicals, and it may occur years before an autoimmune disorder becomes obvious enough to be detected.
One long-suspected culprit in type 1 diabetes is the Coxsackie virus, a common pathogen. It usually causes a mild illness that shows up as passing skin rashes and mouth sores. But Coxsackie can attack the pancreas under the right circumstances, Mallone says: “These viruses can infect beta cells, so they can kill some if there’s enough of a viral load.” That could lead to inflammation that draws more immune cells to the place where beta cells are dying. In their death throes, the betas could release signature self-antigens that sensitize nearby immune cells, causing them to go after other beta cells, because such cells have similar antigen signatures.
That multipart mix is crucial, Mallone says. “Basically you need three ingredients: self-antigen, an inflammatory environment and autoimmune predisposition,” he says. “And these ingredients have to meet at the same place and the same time. This is probably one reason it’s so difficult to identify environmental triggers: because we’re all exposed, but it depends on specific conditions.”
This view is replacing an older idea: that viruses trigger autoimmune reactions when the viral proteins look, molecularly, a lot like self-antigens, leading immune cells to get confused by the similarity and attack the self. Support for this idea, called viral mimicry, began to crumble as researchers amassed evidence that such molecular doppelgängers are fairly common but very rarely cause disease. There are a number of molecules in human bodies and infectious agents that look like one another, and they are called cross-reactive. “Cross-reactivity is extremely abundant; you can find it everywhere,” says DeLisa Fairweather, an immunologist at the Mayo Clinic in Jacksonville, Fla. If cross-reactivity led to illness, she says, “we should see diseases at higher levels. It’s not the answer.” Thus, the idea has gained currency that a virus kills some target cells and creates some inflammation, and in the middle of all this, some immune cells become sensitized to the dying cells’ proteins.
A Rising Count
Other irritants from outside the body, such as drugs and other chemicals, can create these inflammatory conditions, providing more occasions for encounters with immune system sentinels to go wrong. Some scientists think encounters with such substances can explain a spike in autoimmune disease frequency over the past few decades. In 2020 rheumatologist Frederick Miller of the National Institutes of Health and his colleagues published an analysis that tracked the prevalence of antinuclear antibodies, a subset of autoantibodies that go after proteins in a cell’s nucleus. The study followed more than 14,000 participants in the U.S. over a 25-year period. Between 1988 and 1991, 11 percent of those tested carried the antibodies. That number stayed roughly stable through 2004, with a small rise toward the end of that period. But by 2012 the data showed a big jump in the number of people carrying the antibodies, to nearly 16 percent of participants. The rise was particularly striking in adolescents, Miller says, “which to me was the scariest thing.” That could indicate a coming wave of autoimmune diseases.
What has changed to cause the increase? “We don’t know yet,” Miller says. He can, however, rattle off a number of possible environmental and behavioral factors: during the time span of his study, approximately 80,000 to 90,000 new chemicals have been approved for use, he says. “We have a completely different diet,” he continues. “Our use of electronics, while handy and useful, has also meant we’re not getting enough sleep.” People have also encountered increases in air, water and food pollution. “There are hundreds of differences in our lifestyle and exposure in the past 30 years,” Miller says.
Another, somewhat paradoxical idea is that our immune systems now get too little exposure to the outside world and consequently overreact when they encounter relatively benign molecular representatives from it. The theory is related to a notion called the hygiene hypothesis, put forth in the early 2000s, which was based on changes to sanitary conditions. This iteration is more closely tied to changes in our intestinal bacteria, our gut microbiome, that come with the habits of modern society. A quirk of geopolitics has presented a unique opportunity to study this effect. After World War II, a region in northeastern Europe called Karelia was split into Finnish and Russian territories. The populations are genetically similar, but Finnish Karelia rapidly modernized after the war, whereas living conditions in rural Russian Karelia remained similar to those of 60 years ago. You might think an improvement in living conditions would reduce the burden of disease, but that has not happened with autoimmune illnesses. In fact, the reverse effect has been observed. The incidence of type 1 diabetes in Finland is the highest in the world and about six times that in Russian Karelia. Rates of other autoimmune diseases such as celiac disease are also six to 10 times higher in Finland compared with Russia.
Mikael Knip of the University of Helsinki and his colleagues think these changes in disease prevalence can be tied to changes in the microbiome in the Karelian populations. Our internal ecosystem of gut bacteria becomes established during infancy and is heavily influenced by our surroundings. For a 2016 study published in Cell, Knip’s team collected stool samples from children on both sides of the Finnish-Russian border for the first three years of their lives. “When we analyzed the data, we did see a clear difference between the infants in Finnish and Russian Karelia,” he says.
The microbes in Russian children were dominated by a nonpathogenic form of Escherichia coli. Finnish children, in contrast, were home to high levels of a bacterial genus called Bacteroides, which typically does not prompt a strong immune reaction. “Exposure to various microbes in the environment has an effect on training the immune system, particularly in the first year of life,” Knip says, and it looks as if the Finnish systems did not get very rigorous training. The Finnish microbiomes also lacked the variety seen among the Russians. Although Knip emphasizes that it is too soon to cement a cause-effect relation between any drop in microbiome diversity on the Finnish side and the rise in autoimmune problems, he does think there is a connection. “I’d say we need to rename the hygiene hypothesis to the biodiversity hypothesis,” Knip says.
Sharma agrees that gut biodiversity is important, and exposure to variety—for example, by eating nonprocessed foods or at least much less heavily processed versions—is key for training the immune system. “That makes our gut flora more abundant and more diverse,” she says. And it gives our immune system a chance to get used to molecules that are not inherently dangerous so that it does not overreact when it encounters them.
Ultimately a better understanding of the causes of autoimmune diseases—and improved treatments for them—will come from an approach that is quite different from narrow studies of immune cells or target tissues or the microbiome, Miller says. It will come from a more holistic view. Much like in the old Indian parable, he says, “everyone’s looking [at autoimmunity] like the blind men feeling around the elephant, where every person is seeing different things, and they’re not able to connect these to the whole.” Seeing the entire immunological picture will require more studies on different types of biomolecular dynamics, and that research will have to be done in large populations of people. Such an approach could amount to “millions of data points,” Miller says. “We need to embrace the complexity.”

 Rights & Permissions
Rights & Permissions
