
We may well remember the 21st century in two halves: the time before SARS-CoV-2 and the time after. Despite decades of warnings about the potential for a deadly global pandemic, public health systems worldwide were completely outmatched. The first COVID-19 patients were admitted to a hospital in Wuhan, China, in December 2019, and several of them died. Many Americans assumed that even if China failed to contain the virus on its own soil, the span of an ocean would protect them. This complacent view ignored the fact that previous coronavirus outbreaks—caused by SARS-CoV (for severe acute respiratory syndrome coronavirus) and MERS-CoV (for Middle East respiratory syndrome coronavirus)—reached several continents; MERS-CoV has yet to be eradicated. And so SARS-CoV-2 arrived on American shores by early 2020. The public health response was chaotic and varied from region to region. Some city and state governments invoked stay-at-home orders and mask-wearing mandates. Others simply hoped for the best. Similar scenarios unfolded around the world. By early 2022, 5.8 million had died globally.
Despite the disorganization at the national level, medical professionals and research scientists launched an all-out effort to counter the new threat even before it arrived in the U.S. Two years later this global collaboration has generated unprecedented insight into the coronavirus and its impact on the human body. We are beginning to understand why SARS-CoV-2 results in wildly different degrees of illness. Some people exhibit no symptoms; others develop a cough or a fever. Most gravely, some fraction of patients suffer a life-threatening pneumonia and a condition called acute respiratory distress syndrome (ARDS).
Researchers now know that the virus, like SARS-CoV and MERS-CoV, can provoke the immune system to misfire—and the resulting inflammation may lead to ARDS and an array of perilous symptoms. Readily available clinical tests show clear evidence of high levels of immune proteins—interleukin-6, tumor necrosis factor α and C-reactive protein—in the blood of seriously ill patients. A few months into the course of the pandemic, the welcome but limited success of broad immune-suppressing drugs, such as the corticosteroids prednisone and dexamethasone, confirmed suspicions that in the sickest patients the immune system had gone into hyperinflammatory overdrive. These same anti-inflammatory treatments were widely used for severe infections with the preceding coronavirus outbreaks.

We now know that in a certain fraction of COVID patients, an unbridled immune response causes damage throughout the body, producing blood clots, heart damage and even organ failure. The most severe cases require hospitalization in intensive care units. The standard retinue of steroids are not enough for treating severe COVID: these patients will require more targeted treatments. We also badly need rapid tests that can examine tissue samples for biological indicators, or biomarkers, that predict the course of the disease—for example, the likelihood that a patient diagnosed with mild COVID will go on to develop a severe case.
Immunological Misfires
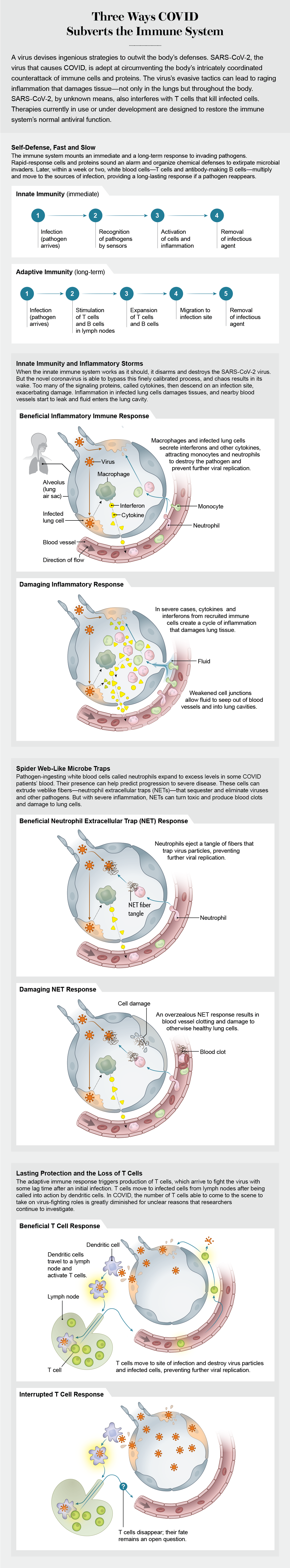
Developing biomarkers and drug treatments requires a deep understanding of how SARS-CoV-2 interacts with cells throughout the body and how the immune system then responds to the virus’s arrival. In the spring of 2020 our laboratory, in collaboration with many others, began to examine the dysregulated immune reactions that underlie severe COVID cases. We knew when we started that the immune system choreographs an intricate chain of events in response to invading pathogens. We also knew that if any of the steps in the immune response are mistimed, it can lead to exaggerated levels of inflammation that damage the body’s own tissues.
The immune system has in its arsenal a fast, emergency response and a slower but longer-lasting defense against viruses, bacteria, fungi and other pathogens. The “innate” immune system acts as a first responder. Some receptors on and inside these immune cells sense invaders, activating an elaborate signaling cascade using proteins called cytokines. The cytokines warn nearby cells to put up defenses, initiate the death of an infected cell or heighten the alarm to bring in other types of cytokines. Innate immune cells also summon certain white blood cells to build more durable immunity to the pathogen. Within a week or two these members of what is called the “adaptive” immune system become active by increasing levels of highly targeted antibodies and T cells that eventually disable or kill an invader.
In most COVID patients, the innate immune system performs as it evolved to, disarming and killing SARS-CoV-2. In a small percentage of cases, however, the body’s counterattack does not proceed as planned. When this carefully timed cascade of signals goes awry, innate immune cells react by making too many cytokines. The overproduction of diverse signaling molecules in COVID resembles “cytokine storms” that turn up in other medical conditions and were thought to be a factor in severe COVID. Research suggests that, in most cases, the inflammation differs from that of a cytokine storm, even though it still poses a threat to patients. It can bring about ARDS, resulting in lasting damage to the lungs or other tissues. It can also lead to the buildup of fibrin, a protein that causes clotting. If this were not enough, it can induce fluid leakage from blood vessels, triggering respiratory failure.
Viruses harness the human cell’s machinery to reproduce themselves. One innate immune system strategy undermines the virus’s ability to multiply, but it appears to falter against SARS-CoV-2. In 2020 researchers devoted attention to a class of cytokines known as interferons, a first line of defense that can block the various steps of viral replication in a cell. Rapid production of type I interferon (IFN-I) by the immune system may enable a virus to be brought under control and check any progression beyond mild disease. But some studies have suggested that in older adults or patients exposed to large amounts of a virus, the immune system may lag in its response, allowing the virus to continue reproducing. Further, when interferons finally do arrive on the scene, they may overreact, spurring the manufacture of high levels of diverse cytokines, which can lead to inflammation and severe illness. Measuring the interferon response may furnish vital knowledge about whether a COVID case will progress to a life-threatening illness, and it may provide clues about how to treat the infection.
The science is still evolving, however, and there are many ways the immune response could run askew. For example, the virus may hamper a person’s ability to make interferons. Alternatively, a given patient might produce less IFN-I because of genetic factors. It is even possible for a person’s immune response to be so erratic that their body makes antibodies against IFN-I. We and others have investigated the presence of these “autoantibodies” as a possible cause of long-term COVID symptoms. Detection of autoantibodies could serve as a useful biomarker to predict whether a patient’s condition will worsen. It was hypothesized that some patients might also benefit from an infusion of lab-made interferon, and clinical trials of such treatments were underway by late 2020.
An inflammatory eruption
Cytokine storms made headlines in severe cases of the previous coronaviruses (SARS-CoV and MERS-CoV), so when SARS-CoV-2 emerged it was natural for scientists to suspect that a similar mechanism was at play. Early in the pandemic, physicians did detect elevated cytokines in patients, but the amount of these proteins and the subsequent inflammatory state they evoked differed from that of a classic cytokine storm.
Whipping around inside these patients were high levels of cytokine proteins that, depending on the cell receiving them, could lead to a range of outcomes, some of them detrimental. Some cytokines, such as IL-6, TNF-α, IL-1β and IL-12, amplify inflammation and tissue damage. Diane Marie Del Valle of the Icahn School of Medicine at Mount Sinai and her colleagues reported significantly elevated levels of some of these cytokines in the blood of nearly 1,500 patients from the New York City area. Findings from this group indicated that abnormally high levels of IL-6 and TNF-α could serve as reliable predictors of disease severity and death.
We saw the same changes in the patients we were tracking at the time. Moreover, our lab and others began to recognize some unusual outliers among patients’ cytokine profiles compared with a typical cytokine storm. We observed high levels of IL-5 and IL-17, cytokines not classically associated with antiviral immune activity. Instead these cytokines initiate a seemingly misguided response—one better suited for infections by parasites and fungi. We have yet to understand whether this response causes damage to tissue or just diverts resources the body needs to fight the virus.
In some COVID patients we also found elevated levels of chemokines, a subclass of cytokines that guide immune cells to where they are needed. High concentrations of the chemokines CCL2, CCL7, CXCL9 and IL-8 generated at infection sites served as a rallying trumpet. Not only were cytokines and other immune messengers causing local damage, chemokines were also calling in cells from throughout the body to join the fray.
To identify the source of tissue damage, a number of research groups decided to look at cells in the blood and lungs. In the field of immunology, we commonly use flow cytometry, a technique that allows us to tag subsets of cells in the blood with fluorescent antibodies. Using these markers, our group was able to detect a sizable shift in the populations of immune cells circulating in patients, as compared with healthy donors. Two innate immune cell types—monocytes and neutrophils—were particularly abundant.
To take one example: In healthy donors, monocytes make up between 10 and 20 percent of peripheral blood mononuclear cells, a category of commonly studied white blood cells. But in COVID patients, we often found that the proportion of monocytes exceeded the normal range by threefold or more.
As an integral component of the innate immune system, monocytes normally patrol the blood and arrive first on the scene to eliminate or sequester pathogens. When they sense a microbial threat, the cells can respond by differentiating into macrophages and dendritic cells—specific types of white blood cells. Macrophages consume pathogens and cellular debris. Dendritic cells identify and flag a pathogen for other immune cells to respond to.
To ensure that the immune system does not overreact, levels of monocytes are usually tightly regulated, but this control is lost in severe COVID cases. In the worst disease outcomes, monocytes and macrophages infiltrated the lungs. When Mingfeng Liao and others at the National Clinical Research Center for Infectious Disease in Shenzhen, China, peered into the lungs of people with severe COVID (by sampling cells in fluid from the lower respiratory tract using a technique called bronchial alveolar lavage, or BAL), they found an abundance of monocytes and macrophages. In agreement with some other findings, both cell types expressed cytokines at levels similar to those found in severe inflammation. Assuming cytokines, largely produced by monocytes and macrophages, are responsible for enhancing all of this damage, interventions that block their inflammatory activity might prevent severe infection.
Our Lab’s Makeover
On March 1, 2020, New York City confirmed its first case of COVID in what would eventually become one of the most devastating series of community-acquired infections in any city in the U.S. Just 80 miles away, we waited in New Haven, Conn., for Yale University to confirm an impending shutdown, which happened on March 18. In many ways, our initial experiences mirrored those of all Americans placed under state lockdowns. Undergraduates were instantly sent home. Postdocs and graduate students were prohibited from working in shuttered laboratories. As coronavirus spread across the globe, university departments full of trained scientists were sidelined—though not for long. Our lives were about to change dramatically.
In the same breath, scientists at all levels in academia laugh about and lament the gossamer-thin line separating our research lives from our personal lives. The arrival of a global pandemic erased that boundary for those of us who chose to shift our research to address SARS-CoV-2. The sudden influx of patients and the urgency of the pandemic eliminated the luxury of planning experiments or leisurely reading papers. We rapidly shifted from investigating the immune response in cancer, herpes and influenza infection to uncovering its role in COVID-19. Of course, home lives were upended as well.
As a lab that specializes in the immune responses against viral infections, we were ready to contribute our insights—or at the very least our knowledge of the immune system and our skills in working with sophisticated lab equipment. In a flurry of activity, our facility began collaborations with doctors, nurses and administrators across multiple schools and departments at the university and the Yale New Haven Hospital. As soon as the first patients started to roll in, we had a chance to contribute to helping understand how SARS-CoV-2 makes people sick.
Our newly formed team, called IMPACT (Implementing Medical and Public Health Action against Coronavirus CT), performed numerous PCR (polymerase chain reaction) tests daily to supplement our area’s capacity for testing suspected cases of COVID. Compared with reports of two-week turnarounds from commercial testing facilities, we were blazingly fast, with samples arriving in the early afternoon and results available less than 12 hours later. Although we did not realize it at the time, this pace was to become the new norm for research in the lab and represented only our first steps in a pivot toward SARS-CoV-2 research.
Academic research typically proceeds at a slower, more considered pace, but doing science in the time of COVID required us to be just as careful in a fraction of the time. Our efforts to pitch in with PCR testing blossomed into a comprehensive study of the immune cell changes occurring in SARS-CoV-2-infected patients. We received batches of patients’ blood daily, and within hours we translated these crimson tubes into hard data. From each patient, we took a sample roughly every four days, and over the course of months we assembled these daily snapshots into a comprehensive record of the immune system’s fight against SARS-CoV-2. Most important, we were learning in real time what turned a bad infection into a lethal one.
At the same time, all across the world, other labs were racing to perform similar experiments. As you can expect, these parallel efforts can generate comparable data and—more often than you would like—conflicting numbers. But when dealing with patients, it is extremely important to validate a discovery from one lab with the work of another and another on top of that.
People are unique and so, too, are their medical treatments, their underlying conditions and other factors outside of our ability to accurately track, such as how much virus they were exposed to. It is a testament to the robustness of our scientific enterprise that researchers around the globe came to similar conclusions.
The pandemic has also changed the way we disseminate our findings. Instead of waiting for months to publish our work in peer-reviewed journals, scientists quickly shared their findings through preprint servers such as BioRxiv and MedRxiv. This enabled rapid exchange of information and ideas in real time and changed academic publication practices overnight. COVID has brought about a fundamental transformation in the way that basic research is conducted. —A.I. and P.W.
If cytokines are in fact major drivers of severe COVID, it would be logical to try to reduce their presence in patients. There are drugs that do this: tocilizumab, for example, is a therapeutic that blocks the receptor where IL-6, a major cytokine, docks. Unfortunately, results of clinical trials with tocilizumab have been mixed when it comes to evidence of improving disease outcomes. A growing number of scientists and clinicians have therefore looked beyond just cytokine storms for a more complete explanation for the damaging hyperinflammatory responses in COVID.
An additional contributor to COVID immune pathology may be a peptide, or small protein, called bradykinin. By reanalyzing lung-fluid data from patients’ BAL samples, Michael R. Garvin of Oak Ridge National Laboratory in Tennessee and his colleagues formulated a hypothesis that bradykinin may, like cytokines, induce an inflammatory response. These “bradykinin storms” can, in fact, be exacerbated by inflammatory cytokines. In excess, bradykinin may lead to massive dilation of blood vessels and many of the surprising symptoms seen in COVID patients, such as cardiac arrhythmias and sudden cardiac arrest. Researchers also began to find massive increases in the production of hyaluronic acid in very ill patients. Aggregates of this molecule can hold remarkable amounts of water. Postmortem analyses of heavily saturated lungs have shown that the combination of these conditions, along with the leakage of fluid from blood vessels, has proved fatal for some patients.
The involvement of bradykinin in COVID requires further confirmation. Direct measurement of the peptide remains extremely difficult. But some success from an exploratory study with icatibant, an inhibitor of a bradykinin receptor, lends weight to the hypothesis that lower levels of the peptide may alleviate severe illness.
Rogue Microbe Traps
Bradykinin also turns up in another inflammatory pathway found in patients’ blood. Neutrophils, which ingest pathogens, can activate bradykinin production. Various labs, including our own, have found neutrophils to be plentiful in some patients’ blood. High circulating levels of a cytokine called IL-8 in people with COVID can attract neutrophils to sites of infection, including the lungs, and assist in increasing the numbers of these cells as well. Crucially, the presence of elevated neutrophils on the first day of hospitalization reliably predicts later admission to the ICU.
Recent papers suggest why neutrophils may be a culprit in COVID pathology. These cells extrude neutrophil extracellular traps (NETs), which consist of webs of DNA, antimicrobial proteins and enzymes that sequester and kill pathogens. Unfortunately, NETs can also damage tissue.
Looking at lung autopsy specimens, Moritz Leppkes of Friedrich-Alexander University in Germany and his colleagues discovered striking blockages of small blood vessels by aggregated NETs. They also observed NETs in the blood vessels of kidney and liver samples. In addition to physical obstructions, NETs can degrade proteins that inhibit blood coagulation, contributing to the high levels of clotting in severe cases. Acknowledging a possible role for these aggregates, McGill University announced a pilot study of a cystic fibrosis drug that clips apart the DNA in NETs.
These varied studies have made it apparent that SARS-CoV-2 turns the immune system against itself. Innate immune defenses—cytokines, monocytes, neutrophils and others—spin out of control. The adaptive immune system also goes off-kilter. One of the most apparent differences between some COVID patients and healthy individuals is the stark loss of T cells, key components of long-term adaptive immunity.
Researchers have observed that T cells from patients with moderate disease behave differently than those from severely ill patients. Normally T cell populations that target a specific invader, or antigen, grow more abundant as a protective measure, but this was not the case in the sickest patients.
There are two types of T cells: those that directly eliminate virus-infected cells and those that coordinate a response to an invader after receiving signals from cytokines. Loss of both types has been observed in hospitalized COVID patients, but it also occurs in other respiratory infections. Diminished cell levels persist, however, for an exceptionally long time—up to weeks in some COVID patients. From research with other respiratory viruses, we know T cells can travel from the blood into infected tissues. Patients with these viruses show elevated levels of chemokines, such as CXCL9 and CXCL10, which direct T cells to infected sites. While we found plenty of chemokines in the blood of COVID patients, we did not find a similar abundance of T cells.
A number of studies have investigated the lungs of people with severe COVID, where the virus has taken up residence. With a genetic sequencing method called single-cell RNA-seq, researchers identified several subsets of immune cells, including a sizable cluster of T cells. But this finding did not provide a full explanation. Neither these lung experiments nor autopsy studies looking at multiple organs could account for the low T cell numbers overall in the blood. It was likely these missing cells simply died, and indeed many research groups have found evidence to support this conclusion.
How then are T cells disappearing? COVID patients had an expanded number of T cells bearing receptors that indicated that the cells were susceptible to an early demise. Another possibility is that the bone marrow might not make enough of the precursor cells that give rise to T cells, which could diminish the pool of mature cells. Studies of aging and of other diseases have established firm evidence that cytokines modulate the bone marrow’s production of T cells. A similar connection has yet to be definitively proved in COVID, despite the presence of the same inflammatory cytokines. Finally, it is possible that the virus itself is directly killing T cells. Testing these competing hypotheses may lead to therapies that can enhance T cell numbers.
Many of the severe immunological manifestations seen in COVID—drastically elevated cytokines, inflammatory cells that infiltrate the lungs, NETs and diminished numbers of white blood cells—appear in other serious viral respiratory infections. SARS-CoV-2 presents its own special challenges. What stands out is its unprecedented spread during the presymptomatic phases and among people who never show symptoms.
SARS-CoV, the virus responsible for the 2003 epidemic, has a relatively late viral peak of 10 days after the onset of symptoms. MERS-CoV’s viral load peaks seven to 10 days after symptoms set in. But SARS-CoV-2’s viral load tops out earlier, possibly even before symptoms begin. The early peak translates to extremely high viral levels before symptoms appear (which for most people happens four or five days after exposure). These numbers mean an infected person can spread significant amounts of virus before feeling even the smallest tickle in the throat.
The wide array of organ systems involved in COVID symptoms also seems unique among respiratory viruses. SARS-CoV-2 can cause loss of smell, brain fog, gastrointestinal problems, blood clots, cardiovascular problems and even “COVID toes.” The virus can also infect neurons in the brain. Among those who recover, tissue damage can linger for months.
These observations may not be entirely surprising. Three cell types that make up blood vessels—endothelial cells, pericytes and vascular smooth muscle cells—wind through every tissue. All of them are studded with the ACE2 receptor, the portal through which SARS-CoV-2 enters cells. They practically lay out the welcome mat for SARS-CoV-2. Making matters worse, cytokine and bradykinin storms can damage tissue made up of these cells.
Even though the earlier coronavirus SARS-CoV uses the same receptor and can cause cytokine storms and ARDS, there are few reports of the sort of serious extrapulmonary injuries caused by COVID. The viruses are an 80 percent genetic match; it is reasonable to suspect that the other 20 percent of their genomes accounts for the differences between them. But a simpler explanation might be that SARS-CoV-2 has infected more than 50,000 times as many people as its eponymous predecessor and has done so before the eyes of the world’s scientific community.
The past two years of discovery and innovation stand as testament to the dedication of scientists and medical professionals. The research and medical communities have never been more united in their efforts—and never before has the transition from lab bench to patient bedside proceeded as rapidly. Long after COVID, this legacy will remain, and these innovations will persist to counter future pandemics.
 Rights & Permissions
Rights & Permissions

