


In 1907 German psychiatrist Alois Alzheimer published a case report of an unusual illness affecting the cerebral cortex. A 51-year-old woman living in an asylum in Frankfurt am Main exhibited symptoms that are all too familiar to the millions of families affected by what is now known as Alzheimer’s disease. There was memory loss, confusion and disorientation.
After the patient died, Alzheimer examined her brain and made a few key observations. First, it was smaller than average, or atrophic, with a corresponding loss of neurons. Next, there were tangles of protein fibers within neurons and deposits of a different protein outside brain cells. For the next 100 years, these two pathological proteins—known as tau and amyloid—were the focus of research into the causes of the disease.
But there was an additional, often forgotten clue that Alzheimer noted in the autopsy. Under the microscope lens, he saw clear changes in the structural makeup of certain nonneuronal cells. Called glia, they constitute roughly half of the brain’s cells.
After being studied by only a small number of scientists since Alzheimer’s discovery, glia have now entered the spotlight. One type, called microglia, is the main kind of immune cell in the brain and may influence the progression of the disease in different ways during both early and later stages. Microglia might also explain the complex relation between amyloid and tau, the aberrant proteins that lead to neuron degeneration and memory loss.
Research in the past decade has identified new molecular risk factors that implicate these brain immune cells in Alzheimer’s disease. Guided by powerful genetic-sequencing methods, we are beginning to gain an understanding of microglia and of the role of the immune system and its inflammatory processes in Alzheimer’s.
Although we have learned a lot about the biochemistry of tau and amyloid proteins, as well as about some of the genetic and lifestyle factors that can influence a person’s risk of Alzheimer’s, there are virtually no treatments to stop or slow the progression of the disease. On June 7 the U.S. Food and Drug Administration approved a new drug, aducanumab, that removes amyloid from the brain. But it is unclear how effective it is in improving patients’ waning cognitive skills. More interventions are still needed. Recent insights about microglia have suggested potential new therapies for the disease, and several are being developed now, with some already being tested in clinical trials.
Plaques, Tangles and Genes
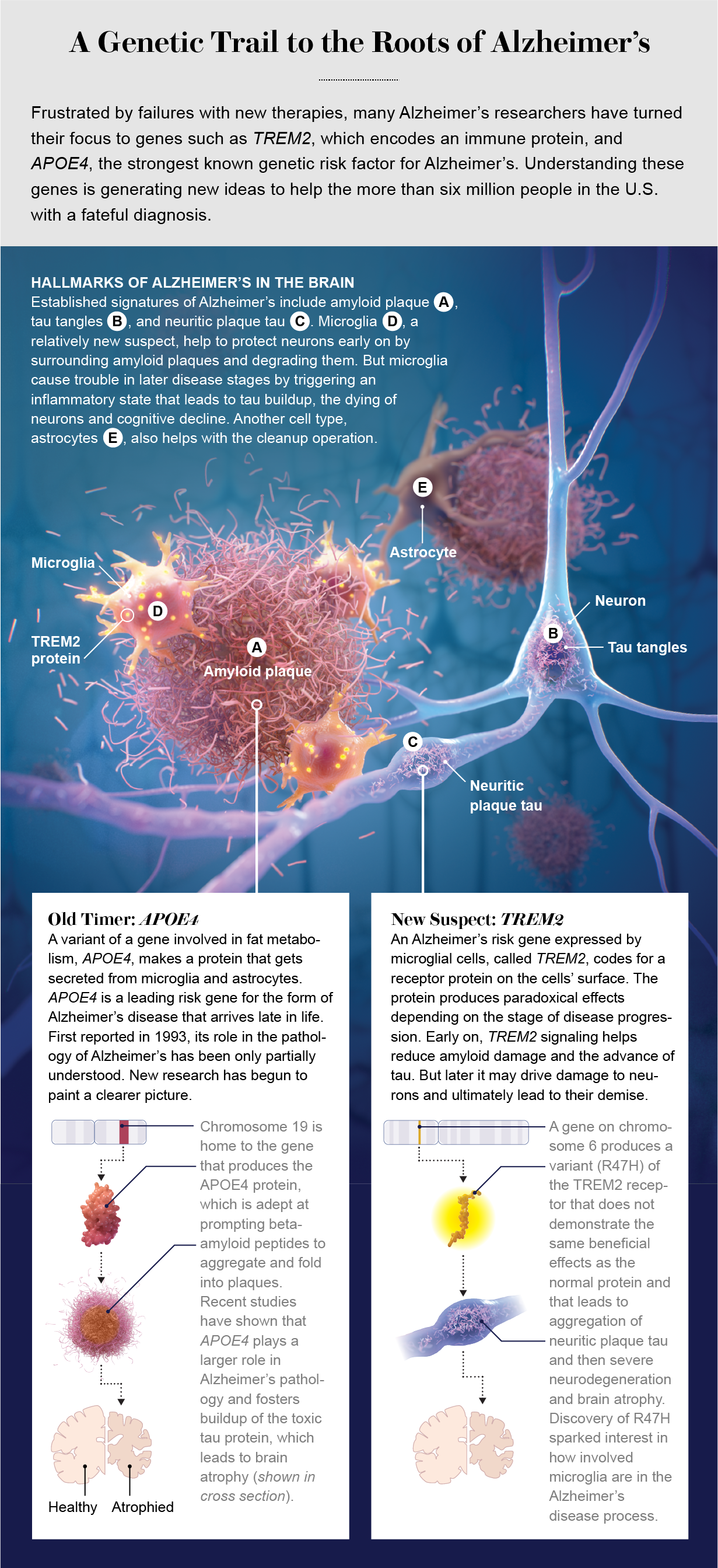
Alzheimer’s disease is the leading cause of dementia worldwide, and its multiple pathologies accumulate and converge over the course of decades. Alzheimer’s has two distinctive molecular hallmarks. The first is plaques made up of one form of amyloid called beta-amyloid. These peptides, or small proteins, are found in the spaces between cells.
The second is the contorted, or misfolded, forms of the tau protein, to which large numbers of phosphate groups get attached in a process known as hyperphosphorylation. This increase in tau phosphorylation has been linked to increased aggregation and toxicity of the protein. Tau is present in twisted clumps, called neurofibrillary tangles, in the cell bodies of neurons. Some tau also turns up adjacent to amyloid plaques in swollen, damaged axons, the long protrusions from the main bodies of neurons. This form is known as neuritic plaque tau.
Both tau and the larger protein from which amyloid is derived have normal roles in cell functioning that get corrupted by the disease process in people with Alzheimer’s. Extensive efforts to understand the pathological forms of amyloid and tau have led to the conclusion that we should consider Alzheimer’s in two stages. The first is a presymptomatic phase of 15 to 25 years during which amyloid builds up in the cerebral cortex, the brain’s outermost layer, in the absence of cognitive symptoms. In the second phase, tau tangles develop in the cortex, and neurodegeneration begins, with cognitive dysfunction appearing as brain cells die.
The Alzheimer’s research community has known for decades that genetic risk factors strongly influence a person’s likelihood of developing Alzheimer’s and that genes provide valuable insight into mechanisms underlying the disease. The foremost risk gene associated with Alzheimer’s is APOE. It encodes the protein apolipoprotein E, which is involved in fat and cholesterol metabolism. (The alphanumeric designations for genes are typically italicized, whereas those for the proteins they encode are presented as regular text.)
The gene’s association with Alzheimer’s, first reported in 1993, relates to one version of it, known as an allele, that dramatically increases the risk of illness. The three common APOE alleles in the human population are APOE2, APOE3 and APOE4. APOE3 is the most common, constituting approximately 78 percent of all alleles, followed by APOE4 at around 14 percent and APOE2 at around 8 percent. Every person has two APOE alleles, and about 25 percent of people carry at least one APOE4 allele. But among those with Alzheimer’s, about 60 percent have at least one APOE4 allele.
People with a single allele of APOE4 have a threefold to fourfold increased risk of developing Alzheimer’s, and individuals with two copies of APOE4 have an approximately 12-fold increased risk compared with people who have two alleles of APOE3. APOE4 carriers have earlier and more abundant deposition of amyloid plaques because their version of apolipoprotein E decreases clearance of beta-amyloid from the brain and facilitates aggregation of the protein. Conversely, people who are APOE2 carriers are at lower risk for developing Alzheimer’s and are much less likely to develop amyloid pathology.
Despite the strength of APOE4’s effect, it does not account for all of the genetic susceptibility to Alzheimer’s. Geneticists have doggedly pursued other risk factors that might explain this “missing heritability,” using advances in gene-sequencing technology to screen thousands of people for changes in DNA associated with a higher or lower risk of developing Alzheimer’s.
Wide-scale screening has identified genetic regions and genes that appear to influence disease risk. Among them are variants of genes—CD33, BIN1, CR1 and MS4A6A—that encode proteins with various functions. For instance, CD33 and CR1 provide the genetic instructions for receptors on the cell surface that detect signals from other cells. These genes discovered by screening across populations have relatively modest effects on disease risk.
Researchers have also sequenced the genomes of thousands of people with Alzheimer’s to look for rare variants that might exert a strong effect on disease risk. Several of these risk genes are expressed predominantly by microglia, the brain’s major immune cells. In 2013 two studies identified a rare variant in TREM2, a gene encoding a receptor that traverses the cell membrane in microglia, as strongly increasing the risk of developing Alzheimer’s.
The sequencing data revealed a variant that substituted an amino acid called histidine for arginine. This mutation was found to impair microglia’s functioning and increase the risk of developing Alzheimer’s roughly twofold to fourfold. Interestingly, like several of the other new risk-factor genes, TREM2 is expressed exclusively by microglia in the brain. These genetic clues suggested that microglia could actively contribute to the disease process, but how?
Surveillance Squad
Microglia are related to immune cells called macrophages that patrol the body to combat pathogens or help repair injured tissue. Researchers are learning that they are involved in everything from defense against infection to pruning excess synapses—the junctions where neurons meet—in the developing brain. Under normal conditions, microglia have small cell bodies with branchlike protrusions that extend throughout brain tissue. The immune cells gobble up—or, more formally, phagocytose—unneeded synapses and debris, and they look for signs of injury or invasion by pathogens.
If injury does occur, the shape and function of microglia change. The cell bodies get larger, and the branches extending from them shorten and decrease in number. Microglia migrate to the site of an injury to initiate an inflammatory response. For decades researchers had observed microglia surrounding amyloid plaques. It was unknown, though, whether they helped to limit amyloid buildup or initiated toxic inflammation. The relation between microglia and tau also was not well understood.
Some studies have indicated that microglia act on neurons to damage axons and synapses, jamming signals transmitted along axons and resulting in an accumulation of tau in cells.
Other research shows that inflammatory proteins called cytokines that are secreted by microglia dramatically increase the destructive process of hyperphosphorylation. The discovery of genetic risk factors such as TREM2 and CD33 pointed to distinct molecules in microglia that could be involved in Alzheimer’s. Researchers hope that understanding how these proteins function will provide insight into the broader role these cells play in the disease process.
Experimental mice are valuable tools for studying how genetic factors can influence the kinds of brain changes seen in human Alzheimer’s dementia. So far there are few experimental animals that reproduce all aspects of Alzheimer’s (for example, a mouse with amyloid-plaque buildup followed by the spread of pathological tau and accompanying neurodegeneration). But researchers have created several dozen “models”—genetically engineered mice that develop either amyloid plaque or tau.
By crossing these transgenic mice with mice engineered to express alterations in Alzheimer’s risk genes, researchers can determine how a gene variant influences different aspects of Alzheimer’s-like pathology. For example, two decades ago amyloid mouse models engineered to express human APOE4 protein were shown to develop more amyloid plaques than mice with the APOE3 or APOE2 gene variants. In recent years researchers have assessed the role of the human TREM2 protein in Alzheimer’s by deleting the Trem2 gene in mouse models of amyloid pathology. Several laboratories consistently identified a dramatic reduction in the number of microglia surrounding amyloid plaques in such mice.
A series of studies from the lab of Marco Colonna at Washington University in St. Louis found that in mice without the Trem2 gene, microglia were unable to properly ramp up their metabolism. When near amyloid plaques, they did not produce sufficient adenosine triphosphate, or ATP, a molecule that fuels cellular activity. Starved of energy, the cells were unable to surround amyloid plaques. Researchers then saw an increase in swollen, injured axons, known as dystrophic neurites, caused by damage from amyloid.
These key observations—fewer microglia surrounding plaques and increased damage to axons—were also seen in postmortem brain sections from people with Alzheimer’s who had a rare mutation in the TREM2 gene known as R47H, the one that had been discovered in 2013. This finding boosted the confidence that the observations in mice could be relevant to how TREM2 works in humans. In addition, work by Jaime Grutzendler’s group at Yale University showed that the fewer microglia that surrounded a plaque, the more damaged the nearby axons were.
That study provided further evidence of the potential role of microglia in protecting against amyloid’s toxic effects in local areas around plaques. It also showed that microglia interact with the ends of tiny amyloid fibers, potentially halting their growth or shielding the surrounding neurons from amyloid’s ill effects.
Toxic Seeds
If microglia protect axons from amyloid plaque damage, might they also safeguard against tau pathology? If so, dangerous TREM2 mutations such as R47H might exacerbate Alzheimer’s pathology by making it easier for neuritic plaque tau to develop near amyloid plaques. Testing this hypothesis remains challenging, but some clues have come from research exploring how tau pathology is able to spread similarly to prions, the proteins that characterize diseases such as Creutzfeldt-Jakob disease, one form of which is associated with “mad cow” disease.
Over the past decade or so researchers have found that tau and amyloid fold up into aberrant shapes similar to a prionlike “seed” that then causes normally structured forms of the proteins to misfold as well. In this manner, pathological tau can propagate to connected brain regions as Alzheimer’s progresses. A series of papers by Virginia Man-Yee Lee’s lab at the University of Pennsylvania showed that injecting normal mouse brains with aggregated tau seeds isolated from brains with Alzheimer’s pathology resulted in the misfolding of mouse tau into neurofibrillary tangles. Mice that already had amyloid pathology developed neuritic plaque tau, the axon-damaging form of the protein. The latter process resembles the chain of events in Alzheimer’s. Although the mice did not develop significant neurodegeneration, the “seeding” approach provided a reliable method to study both amyloid and tau pathology.
When it is not contributing to Alzheimer’s pathology, tau is normally located in the axons of neurons, where it helps to stabilize structural proteins called microtubules that aid in the transport of cellular materials from one part of a neuron to another. Lee’s team found that tau in swollen axons near amyloid plaques became disengaged from microtubules, leaving it potentially more prone to contortion. In essence, these damaged axons turned into fertile soil in which pathological tau seeds in the surrounding amyloid-laden cortex could take root.
Because the detrimental R47H-mutant type of TREM2 protein on microglia increases axon damage, we reasoned that the more common TREM2 form might help the immune cells shield axons near amyloid plaque, preventing tau seeds from spurring further tau accumulation or invading other areas of the cortex. In a study led by Cheryl Leyns and Maud Gratuze, then members of our lab, we injected tau seeds from an Alzheimer’s brain into mouse models with and without genes expressing functional TREM2 protein and found that mice without the protein developed far more neuritic plaque tau in the swollen axons. That damage spread to other regions in the brain through networks of connected neurons.
We also used mouse models developed by Colonna that expressed either normal human TREM2 or the R47H form. Again, amyloid mouse models with the R47H variant developed more neuritic plaque tau pathology near amyloid plaques when injected with tau seeds. To confirm the findings from mouse models, we also examined human brains and found that people with Alzheimer’s-associated TREM2 variants had more neuritic plaque tau. From these observations, we concluded that normal TREM2 and perhaps microglia in general protect against amyloid- induced seeding and spreading of tau throughout the brain.
Microglia seem to be protective against the spread of tau pathology in the amyloid-laden cortex typical of the first phase of Alzheimer’s. But are they still protective once neurofibrillary tangles develop in the cortex and neurodegeneration begins during the symptomatic phase of the disease? Two influential studies—one from Ido Amit’s lab at the Weizmann Institute of Science in Rehovot, Israel, and the other from Oleg Butovsky of Brigham and Women’s Hospital and his colleagues—looked at changes in the activity of microglial genes in mouse models of different neurodegenerative diseases and identified remarkable similarities in how those genes are activated.
They found that microglia in mouse brains with neurodegenerative injuries similar to those that occur with tau pathology switched on diverse genes, many of which encode proteins for degrading unwanted materials in the cell. Microglia at this point strongly increased expression of a mouse version of the APOE4 risk gene. It seems then that both APOE and TREM2 play key roles in determining whether a microglial response is activated when neurons start to die and symptoms first appear.
That discovery led us to cross mice expressing different versions of human APOE with a mouse model that develops both tau pathology and severe neurodegeneration. In a study led by Yang Shi at our lab at Washington University in St. Louis, we found that a tau mouse model that expressed APOE4 had far more neurodegeneration and more advanced tau pathology than mice with APOE3 or APOE2. Next we assessed cell death in people with Alzheimer’s or other neurodegenerative diseases with large accumulations of tau. We found that APOE4 carriers suffered greater damage in the brain than those who carried other alleles.
Additionally, Alzheimer’s patients who were APOE4 carriers declined faster than those who were not. This came as a surprise to us and other researchers because for many years, it had been thought that the primary effect of APOE4 was the accumulation of large amounts of amyloid. These studies, however, pointed to a role for APOE4 not only in regulating amyloid pathology but also in dictating how fast neurons die in response to tau pathology. This implies that the “Alzheimer’s gene,” as APOE4 is known, influences not only amyloid deposition but also neurological damage because of tau accumulation, the two major disease stages.
The expanding understanding of APOE4 led to the next mouse experiment. We found that deletion of the mouse version of the APOE gene was strongly protective against neurodegeneration and delayed the progression of tau pathology—and, more important, the brain damage caused by tau buildup. If deletion of the APOE gene is neuroprotective in mouse models, then perhaps decreasing APOE levels in the human brain would slow down neurodegeneration, particularly in people carrying the APOE4 variant.
In another experiment, we used the tau mouse model, which also expressed human APOE4, to test whether reducing levels of the apolipoprotein variant would protect against neurodegeneration. We collaborated with Ionis Pharmaceuticals to use antisense oligonucleotides, short stretches of modified DNA that degrade messenger RNA (the molecular instructions for a cell to make a specific protein) to reduce the amount of APOE4 in the mice’s brains by half. We found that lowering APOE4 levels when tau pathology was starting to take hold preserved neurons and diminished inflammation and microglial activation in the mice’s brains.
An emerging picture suggests that microglia play two different roles during the progression of Alzheimer’s dementia. In mouse models of amyloid pathology, increased microglial activity around plaques appears to protect the brain. In mice with tau pathology, aberrant tau strongly increases the expression of microglial genes associated with neurodegeneration, and APOE4 seems to further inflame the brain. All of this indicates a strong immune response by microglia to tau pathology that correlates with increased damage, not protection of the brain.
Of course, correlations do not equal causation, and at that point in our research it was unclear whether the strong immune response seen in tau models that expressed APOE4 was driving the degeneration or was simply a response to it. We next asked whether the loss of TREM2, the receptor on the surface of microglia, would increase neurodegeneration and inflammation in the brain. It would not have been entirely surprising if microglia helped to protect neurons, even at that relatively late stage of the disease.
Again, an experiment lowering gene activity was in store. Knocking out TREM2 in tau mouse models decreased the microglial response and diminished neurodegeneration levels. This finding suggested that reducing microglial activity resulted in less damage and brain atrophy from tau pathology.
A Double-Edged Sword
More evidence implicating microglia as drivers of the neuron loss that leads to cognitive decline later in the disease process came from two 2019 studies. Giving mice a drug that blocked the critical protein colony-stimulating factor 1, which microglia need to survive, was shown to remove around 90 percent of microglia in the brain. In tau mouse models, mice that received the drug exhibited dramatically reduced tau pathology and neurodegeneration, indicating that microglia are required for tau-dependent neurodegeneration. These findings demonstrate that TREM2 signaling appears to produce paradoxical effects—either protective or detrimental—depending on the stage of disease progression.
From this research, it seems likely that TREM2 signaling during the presymptomatic and possibly the early symptomatic phases of Alzheimer’s, when amyloid accumulates, helps to reduce the amount of damage amyloid can inflict on nearby axons and synapses. It also impedes the advance of tau through the cortex. Once tau pathology is clearly established, however, microglia may drive synapse loss and the death of neurons.
Assuming that the damaging effect of microglia in mouse models of tau pathology holds true in human Alzheimer’s—still a big assumption—targeting microglia might be a viable treatment strategy. It might be best to promote microglial activation, particularly around amyloid plaques, in the presymptomatic and early symptomatic phases of the illness. Conversely, in more advanced stages of tau pathology, decreasing the microglia response might slow neurodegeneration, as well as the rate of cognitive decline.
Perhaps as we learn more about how microglia behave in response to amyloid and tau pathology, new targets can be identified for the development of therapies to treat this devastating disease. A human clinical trial is currently testing whether TREM2 activation can slow the course of early-stage Alzheimer’s, and multiple other microglia-targeted therapies are entering drug-development pipelines. If these approaches prove successful, it may turn out to be the third, overlooked finding in Alzheimer’s famous autopsy—after the better-known plaques and tangles—that proves instrumental in decreasing the terrible impact on patients’ brains.

 Rights & Permissions
Rights & Permissions
