
No fundamental obstacle prevents us from developing an effective treatment for Alzheimer's disease. Other troubles of human nature, such as violence, greed and intolerance, have a bewildering variety of daunting causes and uncertainties. But Alzheimer's, at its core, is a problem of cell biology whose solution should be well within our reach. There is a fairly good chance that the scientific community might already have an unrecognized treatment stored away in a laboratory freezer among numerous vials of chemicals. And major insights may now reside, waiting to be noticed, in big databases or registries of clinical records, neuropsychological profiles, brain-imaging studies, biological markers in blood and spinal fluid, genomes, protein analyses, neuron recordings, or animal and cell culture models.
But we have missed those clues because for decades we have spent too much time chasing every glossy new finding in Alzheimer's research and too little time thinking deeply about the underlying biology of this ailment. Instead our work has been driven by a number of assumptions. Among those assumptions has been the central and dominant role of the protein fragment called beta-amyloid. A large amount of data supports the idea that beta-amyloid plays an important part in the disease. We have developed drugs that can reduce concentrations of the protein fragments in people with Alzheimer's, yet by and large they have not stopped patients' cognitive decline in any meaningful way.
It now seems simplistic to conclude that eliminating or inhibiting beta-amyloid will cure or treat those suffering from the disease, especially without far deeper and more comprehensive knowledge of how it develops and progresses [see “The Amyloid Drug Struggle”]. We have not been barking up a completely wrong research tree, but our zeal has led us to ignore other trees and even the roots of this particular one.
It is time to go back to basics. I have been a scientist involved in Alzheimer's research for three decades, part of large projects investigating families with a high risk of Alzheimer's, prevention strategies and the physiology of damage to brain cells that is part of the illness. I and my colleagues, who work across many scientific and medical disciplines, believe that we need to reexamine the fundamental physiology and biology of Alzheimer's, as well as reassess the contents of databases and our lab refrigerators for clues that we may have overlooked. This approach will let us develop theories and models of the way this illness progresses, and we can use those ideas to derive novel strategies to combat the disease.
There are at least five potentially fruitful and timely research directions—areas based on important discoveries made in the past several years—that can extend our knowledge, and I believe that they are quite likely to yield insights needed to find effective treatments. These areas range from malfunctions in the way brain cells get rid of problem proteins, to damage caused by inflammation, to trouble with the ways that cells send electrical signals to one another. These are different domains, but in a person they overlap to create illness in the brain, and individually or in tandem they may lie behind the terrible damage done by Alzheimer's.

Protein-Disposal problems
Beginning in the early 1900s, several neuropathologists—including Alois Alzheimer, the scientist after whom the disease is named—described microscopic lesions in the brains of patients who had died with various forms of dementia. Today we know these are clumps of misshapen proteins. In the case of Alzheimer's, some of the clumps consist of pieces of beta-amyloid protein. They sit between neurons and are called senile plaques. Other clumps reside within neurons, made of a protein known as tau, and are called neurofibrillary tangles.
What we still do not know, more than a century later, is why cells fail to remove these abnormal lumps. Cellular mechanisms for the removal of damaged proteins are as ancient as life itself. What has gone wrong in the case of Alzheimer's? This question is as central to the disease process as a loss of control over cell proliferation is to the progression of cancer. Some recent observations from researchers at the Washington University at St. Louis, among other institutions, indicate that abnormal proteins may find their way out of cells, perhaps evading their natural detection systems for bad molecules. We do not know how they do so, but figuring it out might be a very useful way to start a new search for how and why Alzheimer's progresses.
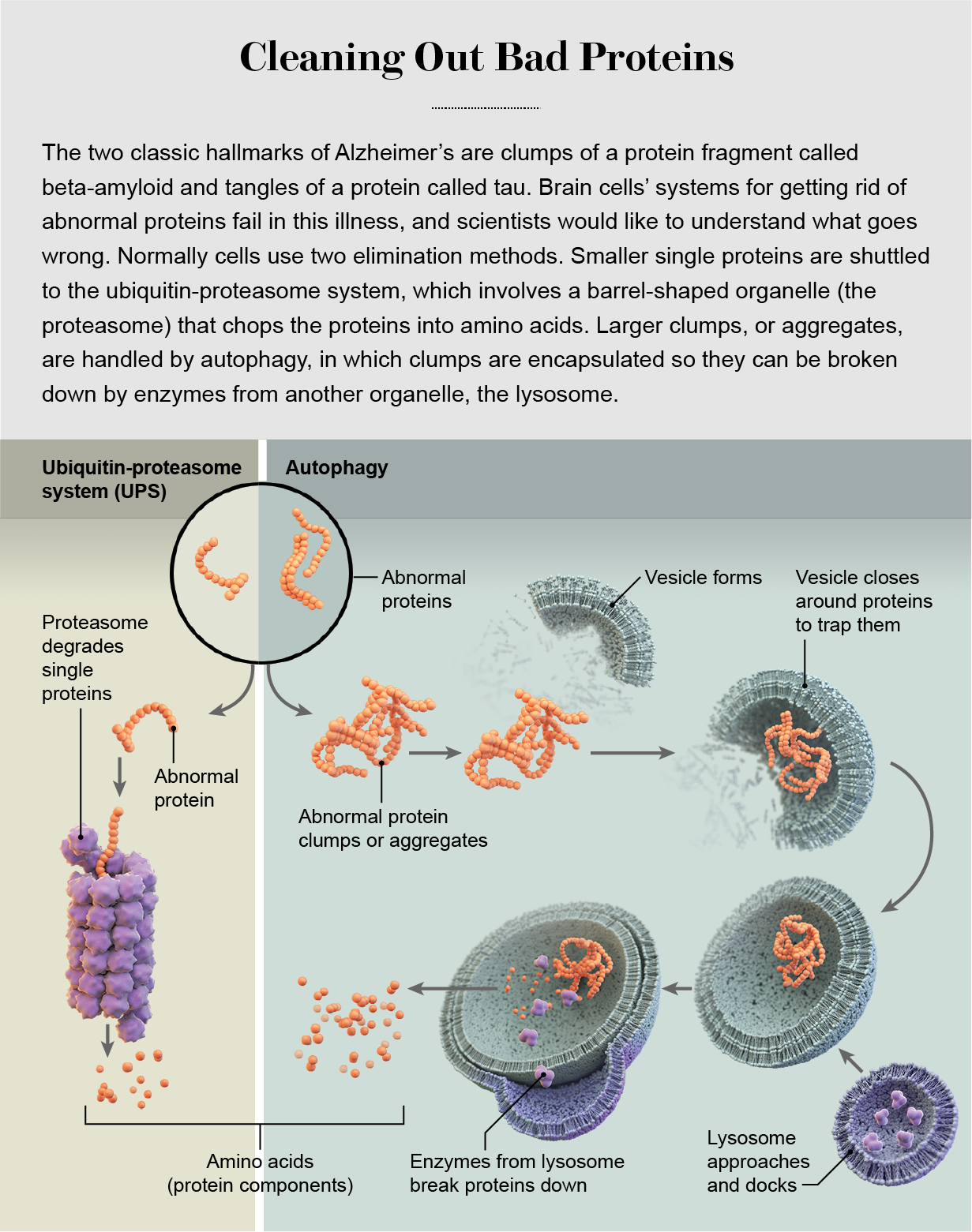
Cells have two major systems for the removal of abnormal proteins: the ubiquitin-proteasome system (UPS) and autophagy. In the former, proteins are inserted into a barrel-shaped cell structure called the proteasome, where they are chewed up into reusable parts; in the latter, the cell wraps up aberrant proteins and totally destroys them. In neurons, these systems are co-opted to control the composition of cell-signaling connections—formed by anatomical structures known as axons, dendrites and synapses—as they are strengthened or weakened during learning. (Sometimes neurons extrude damaged proteins and turn over their destruction to microglia, brain cells that are part of the immune system.)
The decision about whether to shuttle an abnormal protein toward the UPS or autophagy is mainly based on the protein's size. The proteasome has a narrow, pore-like opening at each end that can accept a small, fine, threadlike protein strand. Inside it are enzymes that break the protein down into its constituent amino acids, which are recycled for use in the synthesis of new proteins. Larger molecules that do not fit into the proteasome, such as protein clumps and old, misshapen proteins with age-related damage, get shuttled toward the autophagy system and its more powerful engine of destruction, the lysosome.
In Alzheimer's, something goes wrong and leaves brain cells with these chunks of tau and amyloid that further damage or choke them. So we could learn an enormous amount about the pathology of Alzheimer's if we understood the details of these systems. We need to examine specific differences in the degradation pathways in different subtypes of neurons, as well as the precise mechanism by which these disposal systems recognize abnormal proteins. Malformations in proteins such as tau do not happen in a single step. Proteins may harbor mutations and accumulate modifications that predispose them to misfolding, which can be followed by aggregation into larger and larger structures in a multistage process. As proteins progress along this pathway, at what point do surveillance systems kick in and recognize them as abnormal? In-depth knowledge about these kinds of processes could lead us to a more strategic approach to treatment and intervention with drugs.
One intriguing finding that plays into our understanding of such evasion is that tau can travel out of cells and into the spaces between them, and from there it gets taken up by neighboring cells. What purpose this transit system serves is unknown. Is exchange of the protein among cells normal, or do cells disgorge abnormal tau to rid themselves of a toxic substance? We think that in Alzheimer's, at least some of the tau protein outside cells is already misfolded. We believe this because when such tau enters a neighboring cell, it forms a template, an abnormal pattern, that other tau proteins in that cell use to shape themselves in similar odd ways. When it spreads, tau in neighboring cells copies the specific shape of the entering tau protein.
The observations of tau outside cells have prompted some to speculate that the protein could be intercepted and cleared at that point by an antibody delivered to the patient. But that approach is unlikely to work unless we know exactly how tau is misshapen when it does its damage. This precise structure is necessary information for designing a highly specific antibody. Another open question is where tau resides in the complex space between cells. More specifically, does it move across synapses, where two neurons transmit their signals? This synaptic cleft is a narrow gap that is not easily accessible to an antibody. Potentially more promising approaches are to understand exactly how tau is extruded from cells and the receptors that neighboring cells use to pick the protein up; recent experiments in my lab may point to the identity of one such receptor.
Identifying protein changes
One major recent advance in Alzheimer's research was the imaging of abnormal tau within a cell, snarled in a neurofibrillary tangle, at a level of detail never before seen. This remarkable image, published in 2017 in Nature, showed thousands of tau proteins aligned as pairs tightly locked in a C-shape configuration. It is possible that features seen in this solid inclusion could provide the information necessary to design small molecules that fit within the crevices of the abnormal protein and pull it apart to disrupt the disease process.
But breaking up these structures is a challenging goal for many reasons, not the least of which is how strongly the whole tangle is held together. A more successful direction could be to determine the sequence of microscopic events that takes these tau proteins from their typical liquidlike state to the more rigid and solid state seen in that image and to discover the protein modifications that predispose tau toward this change.
The switch from liquid to solid is called a phase transition. Biologists' interest in such transitions in living cells is now surging because of their possible role in disease. Physical chemists have studied phase separation, such as the condensation of oil drops in water, for many years. Oil and water are both liquids, yet they remain separated because of a balance of attractive and repellent forces. The advantage of phase separation for living cells is that it concentrates a specific set of molecules in one place, which aids certain cellular activities. Multiple proteins near a gene, for instance, can condense to control the expression of that gene, as shown in a 2018 paper in Science. Such a condensed set of proteins, though still in a liquid state, do not diffuse away; they are held together as a droplet by weak physical forces. This configuration allows sets of proteins to move and work together without being wrapped together in a membrane, which would require resource-costly maintenance from the cell.
Some proteins, such as tau, are tightly packed when they are located within a droplet, and the high concentrations could make them prone to aggregation into a tangle. Proteins that form droplets in this way share a property known as intrinsic disorder. Like the Greek god Proteus, they can assume numerous shapes, in contrast to more ordered proteins that are limited to a few specific forms. Different shapes require different energy levels. At times, some intrinsically disordered proteins fold into such a low energy state that they cannot shift out of it, which essentially increases their rigidity. And that may exacerbate their tendency to tangle together.
Cells also pack proteins and other molecules prone to phase transitions in membraneless organelles called stress granules and RNA granules. When certain proteins and RNAs coalesce in such granules, they pack tightly together but typically remain in a liquid state. At a certain density, however, they may become predisposed to more clumping and to a phase change to a solid, a change that would increase their ability to cause brain damage and would make them harder for cell-disposal systems to remove. That is why we need to better understand the conditions that trigger this process.
The Influence of Genes
In middle-aged people, Alzheimer's can arise from genetic mutations in three genes (APP, PSEN1 and PSEN2) that cause a rare familial form of the disease, a frightful inheritance passed from one generation to the next. But the vast majority of the time, Alzheimer's shows up in individuals older than 65 and does not involve these genes. By combing through tens of thousands of genomes, geneticists have now discovered other DNA changes, about two dozen gene variants, that do increase risk by a small amount. The most influential of these alternative forms is a version of the gene APOE known as the e4 variant. A combination of several risk-gene variants adds to one's likelihood of getting the disease. (Because gene variants are frequently associated with ethnicity, we need a much more inclusive data set than the mostly Caucasian-based gene analyses and registries currently available to make a reliable assessment of genetic risk in all populations.)
Each of these variants opens a different door through which we can explore the ways that a small change in our genomes can heighten our likelihood of acquiring Alzheimer's. Some of the more frequently seen variants, and thus the most interesting doors, are genes or other stretches of DNA in the microglia. In a 2019 Science paper examining these immune system cells, scientists found one variant associated with Alzheimer's risk in a gene known as BIN1. This gene is normally involved in the way microglia engulf potentially harmful outside molecules and move them into the cell, protecting nearby neurons. The variant can affect how efficiently microglia clean up stray proteins.
In microglia and other cells, certain gene variants are also associated with age and sex. Differences exist between men and women, for example, for genes on the 22 pairs of non-sex chromosomes and for genes expressed on the X and Y chromosomes. The effects of these variants may have something to do with the higher rates of Alzheimer's in women, which hold even with correction for women's longer life spans [see “The Menopause Connection”]. Overall the small effects of any single gene variant associated with Alzheimer's probably contribute, each in its own limited way, to individual differences in the way we handle amyloid and tau accumulations. We need to nail down the how and why of these contributions.
Taming inflammation
When the brain detects a source of damage such as amyloid plaques or tau neurofibrillary tangles, it sounds an alert and releases a barrage of immune system molecules called cytokines, along with a variety of attack cells. This response stems from the microglia, in large part, and it causes an inflammatory reaction intended to destroy any tissue harboring the trouble spots. This brutelike “innate” system operates quite differently from the more refined “adaptive” immune system, which generates immune cells and antibodies that react only to specific invaders, such as bacteria or viruses, and that mount a narrower, more precise defense. The broader innate response dominates in Alzheimer's. As the lesions proliferate beyond the ability of a neuron's internal machinery to get rid of detritus, this general inflammatory response kicks in and, unfortunately, often hits still healthy cells in the brain. Scientists at the University of California, Irvine, recently have found that eliminating the aged microglia in older mice prompted the animals to repopulate their brains with fresh microglia. This rejuvenation improved spatial memory, reversed age-related changes in neuronal gene expression, and increased the birth of new neurons, as well as the density of their dendrites.
This assault triggered by amyloid and tau probably happens on top of a low level of inflammation in the brain that occurs naturally with aging. Many older people have increased concentrations of proinflammatory cytokines such as tumor necrosis factor (TNF), suggesting that a slight inflammatory state exists throughout the body at this point in life. Aging is highly variable among humans, and the differences mean the progress and the effects of Alzheimer's are quite variable as well. Some of this diversity can probably be attributed to individual variation in human immune systems. Different people inherit distinct configurations of genes involved in immune responses. In addition, during our lives our systems are shaped by nonheritable influences. We get different exposures to symbiotic microbes in places such as our gut and to pathogenic microbes from our surroundings. This all suggests that exposure of the immune system to various pathogens, as well as our genetic differences, may contribute to the way Alzheimer's develops by establishing an individual immune profile, or “immunotype.”
The challenge for researchers who want to stop the brain damage caused by widespread inflammation is to distinguish the desirable immune responses the brain uses to combat developing problems and ordinary age-induced degradation from the other, more reckless immune responses to the advancing pathology of Alzheimer's. The research community would like to tame brain inflammation caused by the disease but does not yet know how to deliver an intervention with precision.
Electrical Disconnections
The brain is an electrical organ: its most defining feature is its ability to encode and convey information in the form of electrical signals passed between neurons, usually by chemicals called neurotransmitters. How Alzheimer's impairs brain cells' signaling and disrupts the way they assemble into functional memory circuits has been insufficiently studied. But now the ability to detect both structural and functional connections is burgeoning thanks to technical advances that allow us to visualize these links in exquisite detail.
Some of these advances involve optogenetics, a way for scientists to stimulate specific neurons in an animal's brain using light. Researchers can offer the animal a reward or fearful experience, then detect which genes become more active. This approach, in an impressive achievement, is now allowing researchers to observe and manipulate specific neurons that encode a specific memory known as an engram, as noted in a 2020 paper in Science. When those cells were stimulated by light alone after the initial experience, the memory of it was recalled. If we can figure out the biology that drives the formation of these electrical memory connections, that information will be crucial in helping us understand how Alzheimer's pathology interrupts this neural circuitry.

Neuroscientists made another advance this year when they discovered that microglia seem to be involved in making the brain forget these engrams by eliminating the synapses that normally connect neurons.
We also know that neurotransmitters are affected in different ways by some of the proteins involved in Alzheimer's pathology. Tau, for instance, accumulates in neurons that use the neurotransmitter glutamate and work to excite signals. But other neurons that inhibit signals—signaling relies on good start-and-stop mechanisms—release a different neurotransmitter, GABA, and are less affected by tau accumulation. The basis for this cellular selectivity and its consequences is unknown, and we need to understand it much better. Scientists have also seen that neuronal activity enhances tau's spread, which could be another important part of the Alzheimer's puzzle.
Not only are signaling cell types affected differently by the disease process, but effects vary in different brain areas, too. For example, areas of the brain related to memory, emotions and sleep are severely damaged, whereas centers related to primary motor and sensory function are relatively spared. One study found that regions of the brain activated when our minds wander, the so-called default or resting state, are the same places where amyloid plaques are first deposited. But we must be cautious in drawing conclusions—a wandering mind does not necessarily cause amyloid deposition.
Sleep is another electrical state of the brain that is increasingly recognized as a factor in the development of Alzheimer's. Levels of both amyloid and tau fluctuate during the normal sleep-wake cycle, and sleep deprivation acutely increases the production of amyloid and decreases its clearance. Deep sleep evokes rhythmic waves of cerebrospinal fluid that may serve to clear toxins, including amyloid, from the brain. Unfortunately, this kind of sleep diminishes with aging. This observation could stimulate work on pharmacological approaches designed to specifically restore deep sleep.
Shared ideas
These research areas are not the be-all and end-all of a rejuvenated Alzheimer's science agenda. There are certainly more. But these five avenues are intertwined and, like biology itself, can be investigated in many cross-fertilizing ways. One hope I have is that as basic science fills in missing information—particularly quantitative information—computational modelers and theoreticians will step in to help predict the impact of Alzheimer's pathology on brain circuitry and cellular pathways. I also would like to see these research directions prompt investigators to think collectively and systematically and to share their ideas in constructive ways. This is how we can come together to push back our ignorance about this terrible disease.
 Rights & Permissions
Rights & Permissions

